Single Molecule Localization Microscopy (SMLM) allows you to overcome the diffraction limit by observing single molecules, thus providing access to the biological environment at the nanoscopic scale.
This strategy relies on the ability to stochastically activate only a subset of fluorescent molecules to distinguish them in space and time.
Indeed, if two light-emitting molecules are too close, their PSF (Point Spread Function) overlap, and it becomes impossible to localize them distinctly, according to the Rayleigh criterion (ref. [4]).
This stochastic activation process is repeated on tens of thousands of frames, then the dedicated SMLM algorithm detects and localizes the individual molecules on each frame, creating in the end a list of molecule coordinates for each frame. A super-resolved image is finally reconstructed from the sum of all these molecule localizations.
Today, there are 3 main methods that allow stochastic activation of molecules: STORM, PALM, and PAINT.
STORM:
STORM stands for STochastic Optical Reconstruction Microscopy (ref. [1], [2], [3], [5], [6]).
It requires organic fluorophores (AF647, CF680, Cy5, CF568, Cy3b…) and a photoswitching buffer, that induces an oxydo-reduction reaction, thanks to two main components:
- A reducing agent (e.g. MEA or BME) to send fluorophores to the dark state
- An enzymatic oxygen scavenging agent to avoid photobleaching
In this buffer, and with sufficient laser power, the fluorophore cycles several times between a fluorescent excited state and a non-fluorescent dark state, allowing the successive localization of molecules. It eventually will become irreversibly bleached.
Advantages:
- High number of emitted photons
A STORM fluorophore emits around 5000 photons, which allows to have a good localization precision (around 10-15 nm), as the localization precision depends on the number of photons:
Where s is the FWHM (Full Width at Half Maximum) of the PSF of the molecule, N is the number of photons collected from the molecule, a is the pixel size on the image plane, and b is the standard deviation of the background (ref. [3]).
- Sample Preparation
The sample preparation for STORM is fairly easy, despite some optimizations: combine your standard immunofluorescence protocol to a published set of STORM dyes (AlexFluor 647 being the gold standard) and STORM buffers (some commercially available).
Drawbacks:
- Fixed cells only
The buffer and the high laser power is toxic for living cells.
- Sample preparation
Finding a good combination of dyes blinking in the same buffer for multi-color imaging can be tricky, though there are methods to circumvent this (e.g. spectral unmixing).
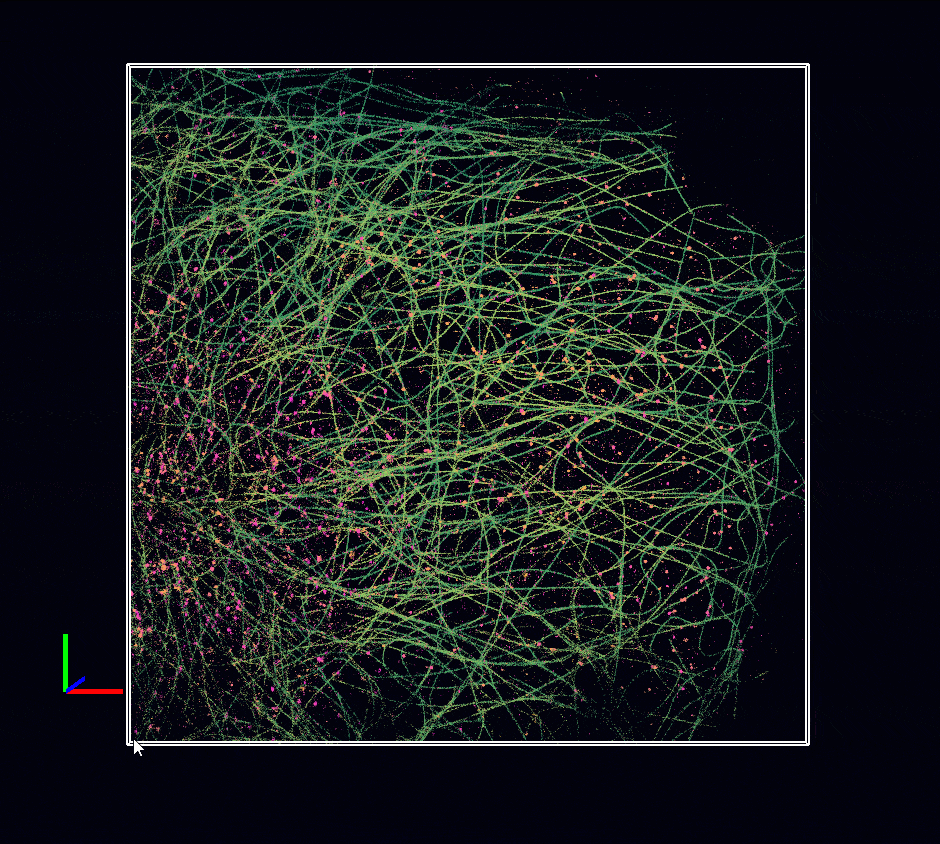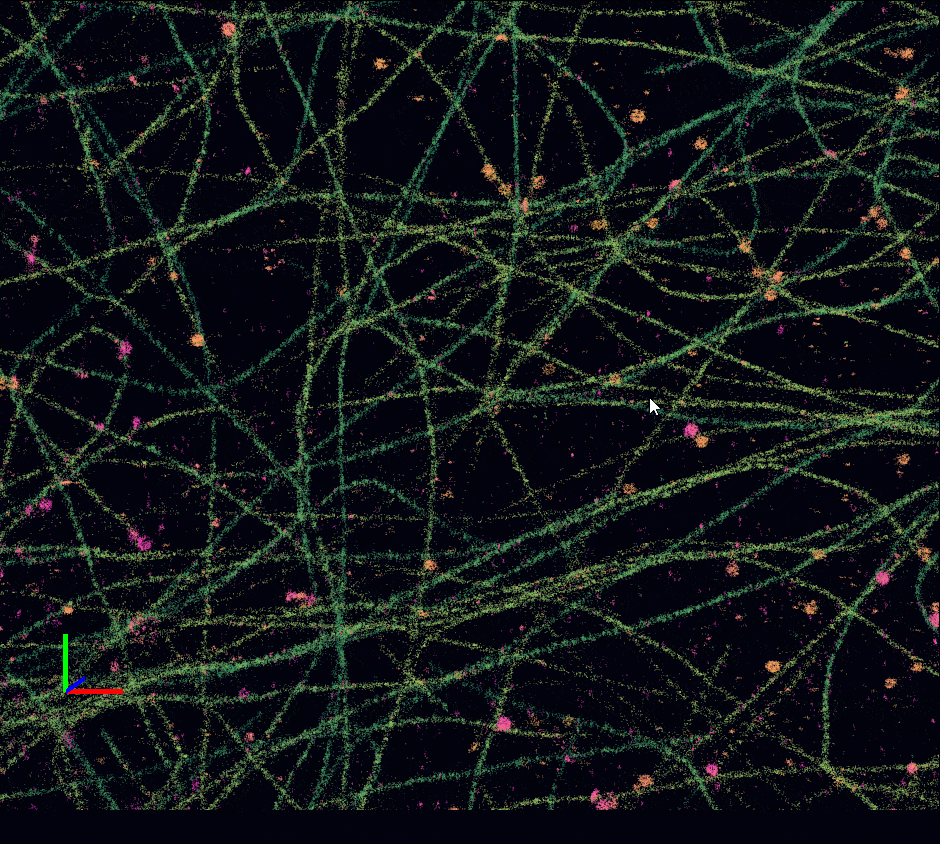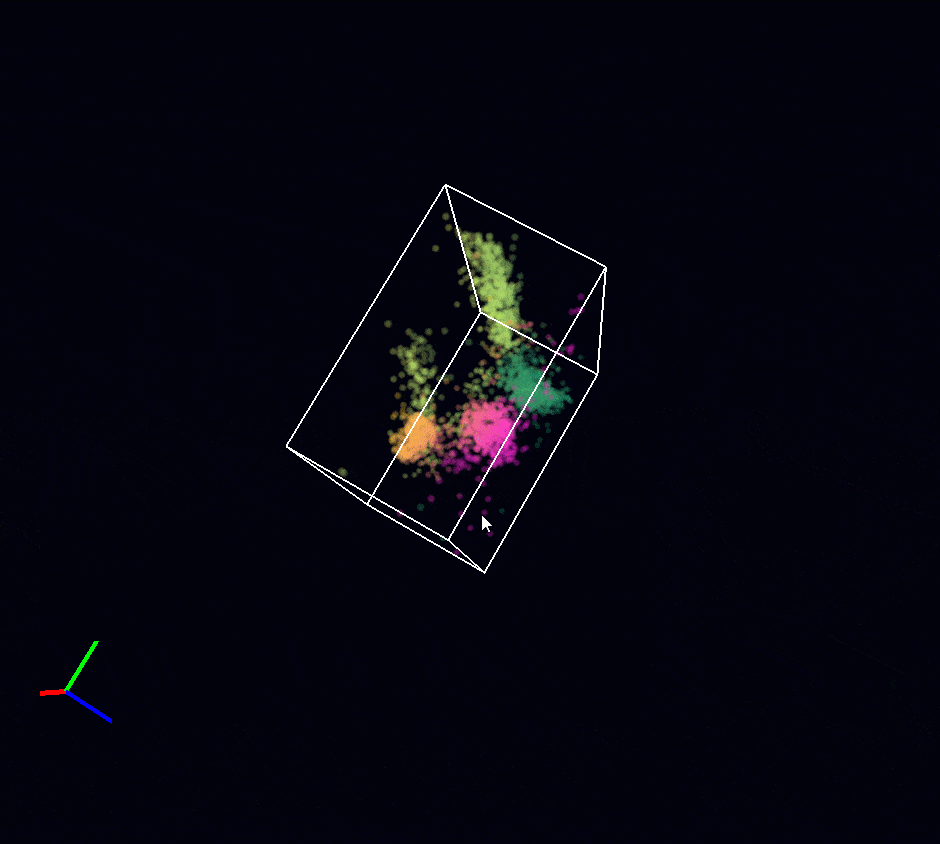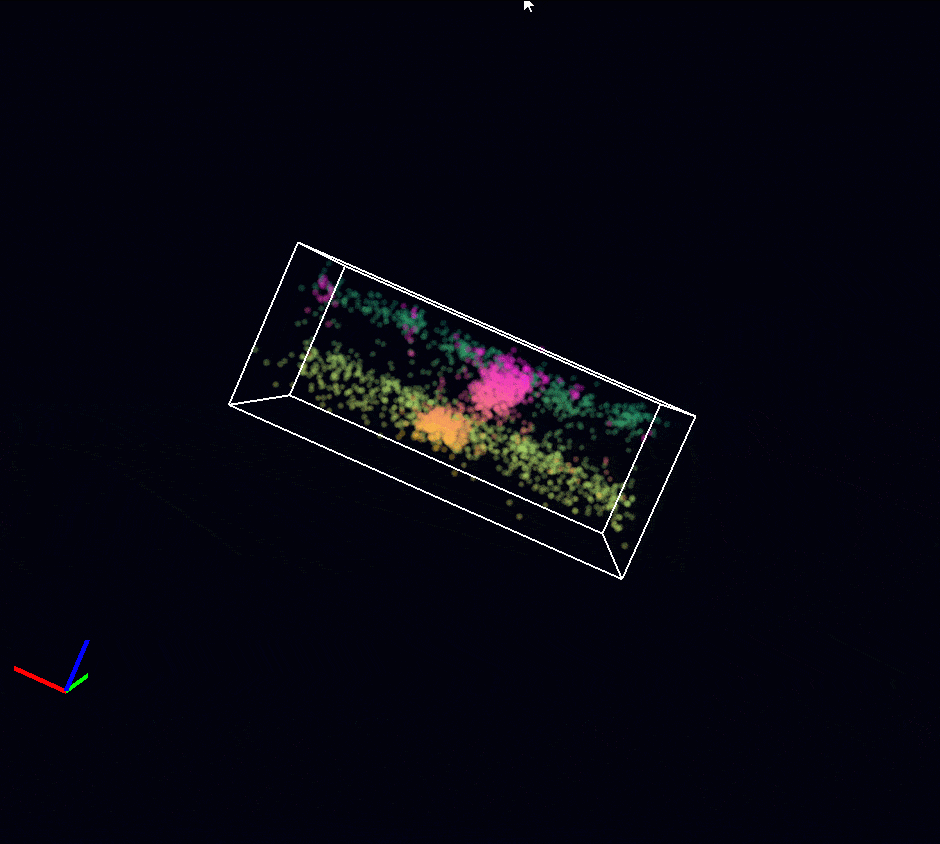

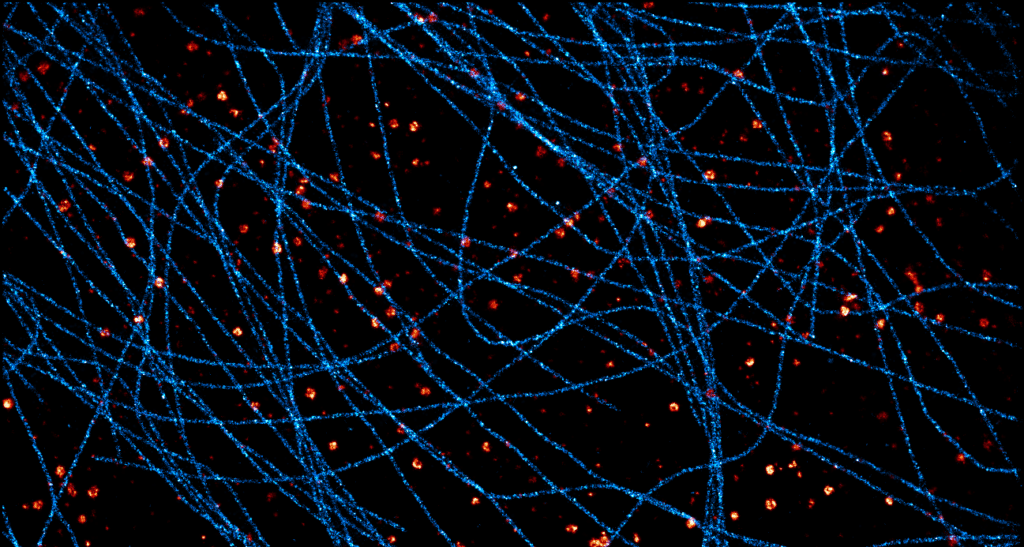
Acquisition: STORM 3D, 2 simultaneous colors
Sample: Cos7 cells – Microtubule (AF647) – Clathrin (CF680)
System: SAFe 360 (abbelight)
Imaging technique: Spectral demixing with a 700 nm dichroic, 3D with astigmatism
Data from Karoline Friedl, NeuroCyto Lab
Acquisition: STORM 2D, 2 simultaneous colors
Sample: Cos7 cells – Microtubule (AF647) – Clathrin (CF680)
System: SAFe 360 (abbelight)
Imaging technique: Spectral demixing with a 700 nm dichroic
Data from Karoline Friedl, NeuroCyto Lab
PALM:
PALM stands for PhotoActivation Localization Microscopy (ref. [1], [2], [3], [7], [8], [11]).
This method requires fluorescent proteins (FP), where a subpopulation can be activated or photo-converted by applying a very weak excitation at 405 nm generally (called the activation laser). A second laser, the so-called “readout laser” at e.g. 532 nm, is then used to excite the activated subpopulation of fluorophores and bleach them after localization.
Fluorophores used in PALM can be divided into four groups : photoactivable (PA; e.g. PAmCherry1, PAmKate, PAtagRFP, PA-GFP), photoconvertible (PC; e.g. mEos, mMaple3, Kaede, Dendra2, PS-CFP2, PSmOrange), photoswitchable (PS; e.g. dronPA, mGeos-M, Dreiklang) and synthetic dyes with self-labeling protein tags (e.g. PAJF-549, PAJF-646).
Their spectral change can be irreversible, or it can be reversible (with PS fluorophores).
Advantages:
- Live or fixed cell imaging
PALM requires low laser powers to activate and excite the molecules and does not require a buffer like STORM (toxic for the cells), which makes it the most suitable solution for live imaging. Notably, it allows single-particle tracking (SPT) on living cells. It is also possible to use PALM to do fixed cell imaging.
- Quantification
By using irreversible PA-FP, it is possible to count precisely the number of molecules in a structure.
- Ability to image any protein.
The use of fluorescent proteins permits to append them to other proteins via genetic engineering and thus allows super-resolution imaging of virtually any protein of interest within the cell, regardless of the availability of antibodies or other affinity reagents.
Drawbacks:
- Lower localization precision
PA-FPs produce fewer photons (~ 500 photons) than STORM / PAINT fluorophores, so PALM generally has a poorer localization precision than the two other techniques. However, recent developments in the synthetic photoactivatable dyes (such as PA-JFs) overcome this problem.
- Labeling
The availability of adequate FPs enabling functionality of the imaged protein can be a limitation, in addition to the known drawbacks of over-expression and complexity of generating endogenously tagged variants. In addition, the efficiency of labeling and expression of the FP in cells or organisms can impair appropriate quantification or imaging.
PAINT:
PAINT stands for Point Accumulation for Imaging in Nanoscale Topography (ref. [1], [2], [3], [9], [10]).
It requires reversibly binding fluorescent probes diffusing freely in an imaging buffer.
Thanks to a chemical affinity, the probes cling transiently to specifically marked sites and can thus be observed during the integration time before detaching from the site of interest, creating a similar “blinking” behavior as used in the methods based on fluorophore photoswitching.
The most common variation of PAINT is DNA-PAINT, for which the labeling is done from the affinity between different strands of DNA. An antibody and the fluorophore are tagged with short, complementary sequences of DNA that will bind and unbind with kinetic properties defined by sequences and buffer composition. These two DNA strands are named docking strand (antibody) and imager strand (fluorophore).
DNA-PAINT buffer is mild compared to STORM buffer and does not change its chemical properties over time. The buffer recipe is simple and can be optimized for best performance of the used sequences. Most importantly – using an open sample chamber – the concentration of the imagers can be adjusted to attain optimal “blinking.”
Advantages:
- No bleaching
Thanks to the large number of imagers in the buffer, it is almost impossible to bleach the sample.
- Good localization precision
It is possible to adjust the length of the DNA-strand to make them bind for more or less time. The shorter the binding time, the faster the acquisition. The longer it is, the more photons are collected, and the more localization precision is increased, but the slower the acquisition goes.
- Multiplexing
Easy multicolor acquisitions are readily available by consecutive rounds of washing and imaging. Thus, it is possible to image a very large number of targets.
Drawbacks:
- Lot of background
Due to the large number of imagers in the solution, there is a lot of out-of-focus light coming from those imagers, creating a strong background. The solution here is to do optical sectioning by illuminating the sample in TIRF (Total Internal Reflection Fluorescence), or by using a Spinning Disk.

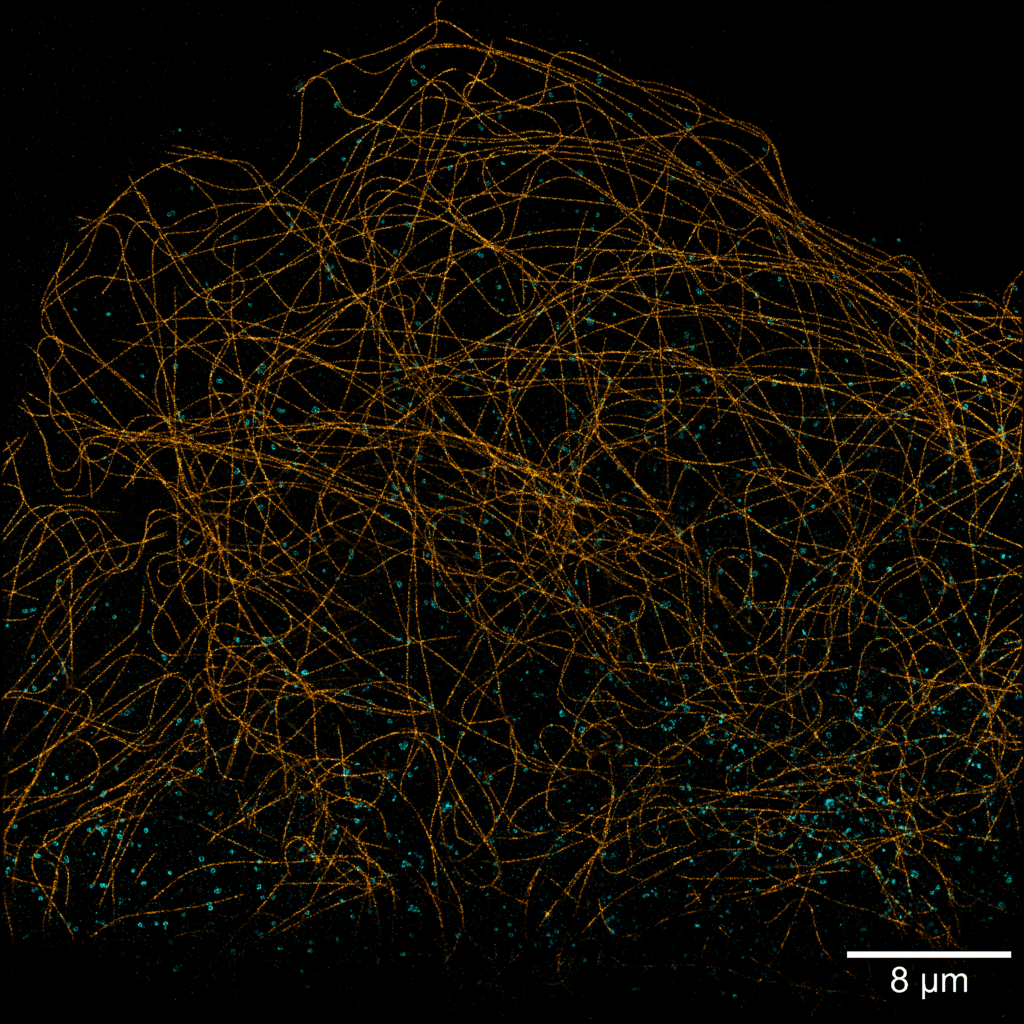
Acquisition: PAINT 2D, 2 simultaneous colors
Sample: Cos7 cells – Microtubule (Atto565) – Clathrin (Atto643)
System: SAFe 360 (abbelight)
Imaging technique: Simultaneous multicolor with a 662 nm dichroic
Data from Karoline Friedl, NeuroCyto Lab Marseille
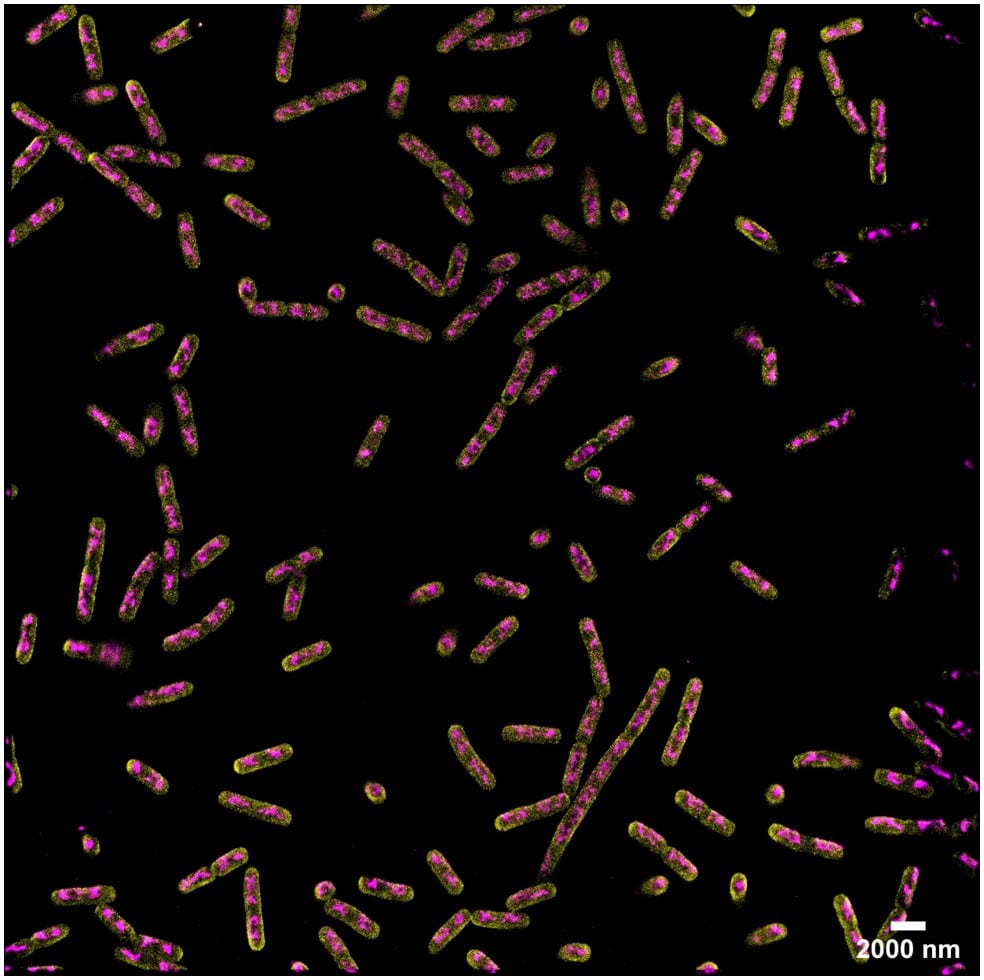
Acquisition: PAINT 2D, 2 simultaneous colors
Sample: Fixed bacteria cells: Escherichia coli – Bacterial chromosome (JF646-Hoechst) – Membrane (Potomac Gold)
System: SAFe 360 (abbelight)
Imaging technique: Simultaneous multicolor with a 50/50 beam splitter + adapted filters
Data from Dr. Christoph K. Spahn, MPI for terrestrial microbiology & Dr. Ipek Altinoglu and Paul Barthelemy, Abbelight
To learn more about abbelight’s single molecule localization solutions, check out the SAFe 180, SAFe 360 and the SAFe RedSTORM.
To see all of the SMLM products that Axiom Optics offers, click HERE.
Sources:
- Pierre Jouchet, Abigail Illand, Guillaume Dupuis, Emmanuel Fort, Sandrine Lévêque-Fort. Dépasser la limite de diffraction en microscopie de fluorescence. Photoniques, EDP Sciences, 2021, pp.44-48. ff10.1051/photon/202110845ff. ffhal-03273149f
- Home | Abbelight
- Single-Molecule Super-Resolution Imaging | Nikon’s MicroscopyU
- Resolution | Nikon’s MicroscopyU
- Rust, M., Bates, M. & Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3, 793–796 (2006). https://doi.org/10.1038/nmeth929
- Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl. 2008;47(33):6172-6. doi: 10.1002/anie.200802376. PMID: 18646237.
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006 Sep 15;313(5793):1642-5. doi: 10.1126/science.1127344. Epub 2006 Aug 10. PMID: 16902090.
- Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006 Dec 1;91(11):4258-72. doi: 10.1529/biophysj.106.091116. Epub 2006 Sep 15. PMID: 16980368; PMCID: PMC1635685.
- Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci U S A. 2006 Dec 12;103(50):18911-6. doi: 10.1073/pnas.0609643104. Epub 2006 Dec 1. PMID: 17142314; PMCID: PMC1748151.
- Schnitzbauer, J., Strauss, M., Schlichthaerle, T. et al. Super-resolution microscopy with DNA-PAINT. Nat Protoc 12, 1198–1228 (2017). https://doi.org/10.1038/nprot.2017.024
- Turkowyd B, Virant D, Endesfelder U. From single molecules to life: microscopy at the nanoscale. Anal Bioanal Chem. 2016;408(25):6885-6911. doi:10.1007/s00216-016-9781-8
- Schermelleh L, Ferrand A, Huser T, Eggeling C, Sauer M, Biehlmaier O, Drummen GPC. Super-resolution microscopy demystified. Nat Cell Biol. 2019 Jan;21(1):72-84. doi: 10.1038/s41556-018-0251-8. Epub 2019 Jan 2. PMID: 30602772.